
<section style="padding: 10px; background-image: none;" border-box;"="" 640?wx_fmt="gif");box-sizing:" 3e8uiaptsaxt8oqeviwtraxjzs8xiliakraax5ya1skxieubva05wbf4oetcgds2jdrvzlehcfrsndaibly8bqianq="" mmbiz_png="" mmbiz.qpic.cn="">Sanger测序在过去的二十年间是基因组研究的主流方法,取得了包括人类基因组计划(human genome project, HGP)等一系列重大成就,这使得单基因遗传病的鉴定和靶向治疗成为可能1,2。随着基因组大数据时代的到来,尽管Sanger测序是基因检测的金标准,但是NGS)技术应运而生,它能够在短时间内产生海量数据3,4,目前二代测序仪平台主要为Illumina和Thermo Scientific。Illumina公司测序仪采用边合成边测序的方法,基于可逆终止化学反应原理,检测测序时产生的荧光信号,而Thermo Scientific公司平台测序仪基于半导体测序原理,检测测序时产生的电信号。两种平台各有优缺点,在临床不同的需求中得到应用。
NGS已经应用于临床许多领域,在辅助生殖,如非侵入性产前检查(non-invasive prenatal test,NIPT);遗传性疾病,如遗传性致病突变筛查;肿瘤研究,如早期诊断,用药指导,预后判断等中发挥着越来越重要的作用。柳叶刀发布的中国疾病负担研究显示,肿瘤和心血管疾病占比高且患病人群巨大,给家庭和国家造成了巨大的经济负担,急需新的检测手段精准指导临床诊疗5。此外,感染性疾病因病原体种类复杂并且传统方法鉴定周期长且困难,也急需高通量的检测方法。本文针对目前二代测序技术除了NIPT之外应用较为广泛的领域:肿瘤、心血管疾病和感染性疾病中应用进展进行了总结。
NGS可应用于全基因组测序(whole genome resequencing, WGS)和全外显子组测序(whole exome resequencing, WES),可获得点突变、小片段插入或缺失、拷贝数变异和结构变异的信息。全转录组测序方法(RNA-Seq)不仅可以检测基因表达谱,还可以检测可变剪接,RNA编辑和融合转录本。此外,可以使用包括染色质免疫沉淀测序、甲基化分析测序等方法研究表观遗传变异。同时研究人员利用强大的生物信息学工具,破译大量数据,以提高我们对疾病的理解,制定个性化的诊疗策略。
1.基于NGS进行多种癌种的研究:在过去的几年中,很多基于NGS的研究已经开展,以提供癌症的综合分子景观,识别有助于肿瘤发生,进展和转移的新的遗传改变,并研究肿瘤的复杂性、异质性和肿瘤的进化。在乳腺癌6-13,卵巢癌14,结直肠癌15,16,肺癌17,肝癌18,肾癌19,头颈部癌症20,黑色素瘤21,急性髓性白血病22,23等癌症中均取得了研究成果(表1)。
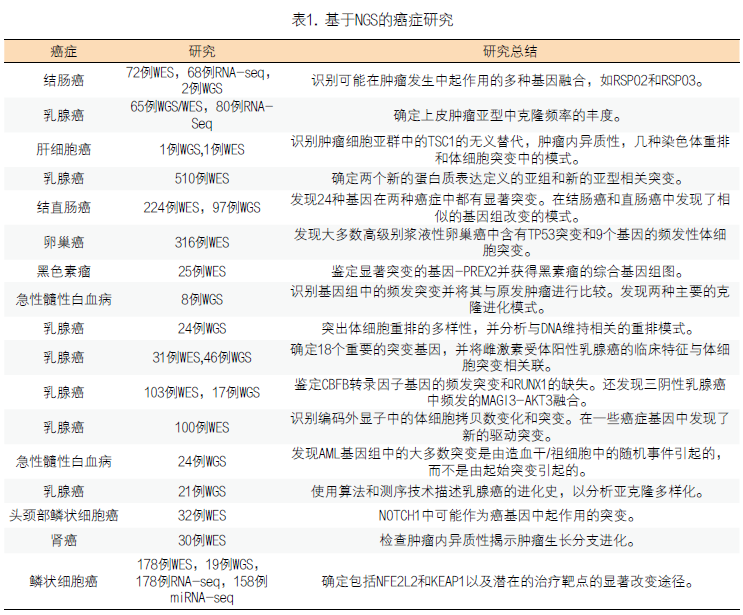
2.识别新的癌症相关基因:癌症主要由遗传突变的累积引起,遗传突变可以在胚系中遗传或在体细胞突变中获得。原癌基因、抑癌基因和DNA修复基因的改变使细胞逃避生长和调节控制机制,导至肿瘤的发生24。癌细胞也可能进一步突变,导至克隆性扩增25。随着克隆性扩增的发展,癌细胞最终会侵入其周围组织并转移到原发肿瘤的远端区域26。癌症基因组的测序识别了很多新的癌症相关基因,特别是在乳腺癌中。有6篇文章报道了他们对大型乳腺癌数据的研究结果:TCGA(癌症和肿瘤基因图谱计划)对来自507名患者的510份样本进行了外显子组测序6,Banerji对103个样本进行外显子组测序,对46个样本进行了全基因组测序8,Stephens对100个样本进行了外显子组测序,Shah对65个样本进行全基因组/外显子组测序,对80个样本进行RNA测序12,Nik-Zaina对21个肿瘤/正常对照样本进行全基因组测序13。除了确认TP53,GATA3和PIK3CA中的频发性体细胞突变外,这些研究还发现了新的癌症相关突变。尽管新的突变发生频率低(<10%),但是特定基因的突变发生在乳腺癌的不同亚型中,并归属于不同的通路。例如,MAP3K1的突变经常发生在Luminal A型乳腺癌患者中6,8,涉及p53,染色质重塑和ERBB信号传导通路的基因在乳腺癌中突变频率较高12。此外,一些突变可以用作治疗位点,如GATA3的突变可能是芳香酶抑制剂疗效的阳性预测标记物8。基因组的测序还有助于完善结直肠癌的突变谱。例如,对72对肿瘤/正常组织配对样本进行外显子组测序鉴定出36,303个导至蛋白变异的体细胞突变。对显著突变基因进一步分析归纳出23个候选的癌症基因,包括KRAS,TP53和PIK3CA,以及新的基因,例如调节细胞周期检查点的ATM。RNA测序鉴定了频发性的R-spondin融合,可能加强Wnt信号传导并诱导肿瘤发生16。另一项研究对224对肿瘤/正常组织进行外显子测序,该研究鉴定了超突变癌症中的15个高度突变的基因和非超突变癌症中的17个高度突变基因。在非超突变的癌症中,除了已知的APC,TP53和KRAS突变之外,还检测到SOX9,ARID1A,ATM和FAM123B中的新的频发突变。对SOX9,ARID1A,ATM和FAM123B的突变和功能作用的分析表明它们是高度潜在的结直肠癌相关基因。并发现非超突变的结肠癌和直肠癌在基因组变异中具有相似的模式。最后对97例肿瘤样本的全基因组测序识别了频发的NAV2-TCF7L1融合15。3.深入揭示肿瘤的异质性和进化:使癌症成为一种难以治愈的疾病与每个克隆中选择和遗传不稳定性导至的肿瘤进化和异质性有很大关系27。这设想最初由Peter Nowell作为癌症的克隆进化模型于1976年提出,试图解释一段时间内肿瘤侵袭性的增加。其它研究人员在20世纪80年代研究了来自小鼠肉瘤细胞系的转移性亚克隆从而进一步支持了这一理论27。NGS的广泛应用揭示了肿瘤异质性和肿瘤进化的机制。通过不同的形态学表型、表达谱和突变以及拷贝数变异,将肿瘤分成不同亚型28-31。在TCGA和Eillis的研究中发现mRNA表达亚型与体细胞突变有关6,8。NGS检测出的大量的体细胞突变展现出个体肿瘤的独特性,每个都包含不同的突变模式。例如Stephens在100位乳腺癌患者中发现73种不同的癌基因突变组合可能10。肿瘤内异质性可以阐释为单个肿瘤内的非同一性细胞克隆或亚克隆,提示不同的组织学,基因表达,转移和增殖潜力。高灵敏度的优点使NGS成为研究肿瘤内异质性的有力工具。一项基于NGS的4例患者肾细胞癌研究成功地阐明了肿瘤内异质性19。对于患者1,在原发肿瘤的9个区域中发现的128个经验证的突变中,40个普遍存在,59个在一些区域共有,29个在特定区域是独有的,表明肿瘤异质性存在并且是“持续的区域克隆进化”19。最重要的是,该研究表明,单个肿瘤活检仅显示肿瘤突变的一小部分;单个活组织检查中,可检测到该肿瘤中所有突变的55%,并且大多数肿瘤区域约有34%的共有突变。癌细胞的持续和平行进化可以建立和维持肿瘤内异质性。例如,在前列腺癌肿瘤细胞水平观察到显著的空间,细胞内和细胞间异质性。在不同患者中观察到单克隆起源和多克隆模型32。肾细胞癌研究中患者1和2中肿瘤区域的系统发育关系揭示了肿瘤的分支进化而非线性进化19。研究还显示了乳腺癌进化的分支结构27。根据“肿瘤生长的树干-分支模型” 27,促进肿瘤生长的体细胞突变,代表了肿瘤发展早期树的树干。这些体细胞突变很可能在这个阶段无处不在。随着时间的推移,其他被称为驱动因素的体细胞突变会导至肿瘤异质性发生,从而导至肿瘤和转移部位发生分支进化。之后,这些分支将进化并变得更加孤立,导至“瓶颈效应”,继而使得染色体更加不稳定,肿瘤异质性进一步扩大27。这致使肿瘤在不断变化的环境中产生适应和生存的能力,并影响药物治疗的疗效19。因此,重要的是检查肿瘤克隆结构并识别位于系统发育树的树干中的常见突变,这可能有助于理解靶向治疗抗性并发现更强大的治疗方法。二代测序在肿瘤中的一些研究成果已经应用于临床,为患者早期筛查、预后判断和靶向用药提供了有力的证据。二代测序在肿瘤精准医学诊断中的应用专家共识也陆续出台33,随着二代测序技术的不断发展和对癌症研究的不断深入,二代测序技术将为癌症治疗的突破提供新的希望。
1. 二代测序在心血管疾病研究中的应用潜力巨大:NGS已被证明可成功鉴定出单基因疾病和心血管系统常见疾病的新型致病突变34。NGS在常见心血管疾病(cardiovascular diseases, CVD)中正变得越来越重要,因为与仅提供已知单核苷酸多态性(single nucleotide polymorphism, SNP)数据的全基因组关联研究(genome-wide association study, GWAS)不同,它可以提供更多信息,包括常见和罕见变异,插入缺失和删除,和拷贝数变异(copy number variants, CNV)35。NGS提供了对大量基因进行并行分析的机会,这些基因可能有助于增进我们对诸如CVD之类的复杂疾病的认识。在鉴定较小家系中的稀有变异时,它也是一种有用的方法。心血管领域中孟德尔疾病的例子包括家族性高胆固醇血症,肥厚性和家族性扩张型心肌病和离子通道病(即Brugada和长QT综合征)36,而临床实践中最常见的CVD则更为复杂,例如冠状动脉疾病(coronary artery disease, CAD)和中风是由复杂的因素导至的基因-基因和基因-环境相互作用37。基因检测不仅被用作研究工具,而且由于其有潜力为家庭提供更多个性化和信息性的咨询服务,因此最近也进入了临床诊断领域38。2. 二代测序揭示单基因心血管疾病中的致病变异:NGS的进展正在改变我们对杂合性家族性高胆固醇血症的流行性评估和对多基因效应的认识。最近,Wang等39发现,靶向NGS检测的导至单基因突变存在于近一半的严重高胆固醇血症的个体中,定义为LDL-Cl> 5.0 mmol/L。当包括拷贝数变异和具有极端多基因得分的个体时,具有单基因突变的个体的百分比增加到53.7和67.1%。在2011年,Meder等人40首先采用靶向NGS方法筛查遗传性心肌病患者。他们采用了含有47个基因的组合来检测10例肥厚性心肌病和扩张性心肌病患者的血液,并发现了27个新的可能有害的突变。在另一项WES研究中,对13个亲子后代和112个非综合征性房室间隔缺损随机个体进行了研究,研究人员确定了新的与先天性心脏病相关基因NR2F2的几种致病变异。Sakai H41使用了两种不同的技术,即重测序阵列技术(ResAT)和NGS,分析了70例非综合征患者(35例胸型,30例腹型和5例胸和腹型)中与综合征性主动脉瘤和/或夹层相关的8个基因。在2011年发表的一项研究中,使用Illumina GA平台,分析了来自HapMap CEU人群的47个欧洲血统随机个体中9p21染色体上大约240 kb区域的数据42。在迄今为止最大的一项外显子组研究中,在来自更广泛的4204个随机个体的外显子组样本中的496名缺血性中风患者中,发现对氧磷酶-1基因的七个变异与缺血性中风有关43。此外,旨在生成和存储WES数据的国际项目,如英国基因组学100,000染色体基因组计划44,将可用于鉴定对卒中的病理生理影响很小但意义重大的多种变异。3. 二代测序揭示具有复杂特征心血管疾病中的遗传背景:NGS技术还用于其他具有复杂特征的CVD相关疾病,包括血栓形成,高血压和血脂异常。在有血栓形成记录的家庭中,NGS已通过将Illumina平台应用于引起遗传性血栓形成的多基因风险检测并获得适当的药物治疗45。在这项研究中,鉴定出200个变异,并使用HapMap不同人群对其进行了评估。此外,正如Costa等人所指出的,多种基因与多种心血管疾病的关联,高血压是中风和冠心病的主要危险因素,占所有心血管疾病死亡率的50%,其遗传因素被认为占30%到50%46。最后,萨达南达等人结果表明,NGS方法可以可靠,准确地检测大量低水平高密度脂蛋白胆固醇患者的致病变异-这是最常见的脂质异常之一,也是CVD的关键危险因素47。通过对患者的整个外显子组(或基因组)进行重测序,揭示了在孟德尔疾病研究中NGS可能是有效的方法48。此外,将这些技术应用于复杂疾病,包括CAD和其他CVD,可能会更深入了解这些疾病的遗传背景,可以实现检测结构变异和罕见变异,以及将不同类型的变异与表型联系起来的挑战。2009年,美国国家心脏,肺和血液研究所(NHLBI)的外显子组测序项目(ESP)成立,目的是发现与心脏,肺和血液相关疾病有关的稀有蛋白质编码变异。为了确定与CVD相关的罕见变异,使用外显子组测序来诊断心血管队列的良好表型47。如前所述,WES和WGS已鉴定出许多在孟德尔疾病和复杂CVD条件下均重要的罕见变异。尽管WGS的覆盖范围比WES好(外显子组几乎占整个基因组的1%),但由于成本高和序列读取深度低,在大量个体中使用WGS仍不可行,WES可能一种中间可行的方法。WGS绝对是进行基因检测的最佳选择,尤其是在心血管疾病中。由于非编码变异在增加CVD风险中起着显著作用,并且测序成本不断降低,因此在将来,WGS将比WES具有更高的成本效益。WGS在揭示未知遗传疾病的病因以及诊断具有非典型表现的已知疾病的患者中更有用。此外,可以通过使用功能基因组学方法(例如RNA-seq和ChIP-seq)来开辟检测疾病相关的生物学途径的新方法。除了确定这些疾病的环境危险因素外,阐明CVD的遗传背景还为精确有效的治疗提供了新方法,NGS技术为个性化医学的发展指明了道路。在心血管医学中,NGS已被证明可以成功地识别新的致病突变,并可以诊断由单个基因中的单个变异体引起的孟德尔疾病。NGS提供了以无偏见的方式(即不了解潜在的生物学机制)对大量基因进行并行分析的机会,这可能有助于增进我们对诸如CVD之类的复杂疾病的病理学的认识。
在过去的五年中,NGS的应用已从研究工具过渡到诊断方法,并且在临床微生物学实验室中变得越来越普遍。这些应用包括(1)全基因组测序(WGS),(2)靶向二代测序(tNGS)方法和(3)宏基因组二代代测序(mNGS)。1.全基因组测序在感染性疾病中的应用:WGS应用于微生物基因组的测序和组装。迄今为止,WGS的最普遍用途是鉴定,分型和/或预测微生物病原体的药敏性。通过识别和跟踪疫情,WGS已被证明在医院和公共卫生流行病学研究中具有重要的作用。例如,WGS能够检测和监测产生CTX-M-15的肺炎克雷伯菌克隆和产生大肠粘菌素的碳青霉烯酶的肺炎克雷伯菌的传播,从而指导感染,控制干预措施并防止这些多药耐药生物的进一步传播49。WGS用于从新生儿重症监护病房的患者中分离腺病毒基因组从而有助于识别和遏制疫情暴发,并且比通过Sanger测序进行分型具有更好的分辨率50。此外,WGS用于检测腺病毒阳性环境样品,从而确定感染的途径,并导至感染防控方式的改变50。除了医院的流行病学和感染控制应用之外,WGS还支持公共卫生计划,以迅速发现,应对和阻止病原体的传播51,52。WGS允许对潜在暴发菌株及其相关性进行更深入的探索,从而使人们对暴露和传播的途径有了更好的了解53,54。除了识别和跟踪暴发,WGS还提供有关病原体毒力和新的耐药机制检测的未开发信息。可通过鉴定毒力因子基因来研究毒力,目前,毒力因子基因尚未被临床实验室检测到,也未用于患者的治疗和管理决策中。例如,一项研究强调了其在金黄色葡萄球菌中检测和分选某些毒力基因(例如 spa和PVL毒素)的潜在实用性55。此外,WGS可以提供对耐药新机制的早期检测,而传统的分子检测方法(例如特定基因或基因座的聚合酶链反应(PCR)可能会错过这种机制)。WGS在检测新突变中的价值的一个例子是在结核分枝杆菌中发现 Rv3792的同义突变,该突变可通过增加下游embC基因的表达从而导至乙胺丁醇MIC升高56。这与上述应用相似,WGS还可以提供对病毒病原体及其耐药性的更详细分析。尽管目前可以通过Sanger测序对一些病毒病原体(主要是人免疫缺陷病毒(HIV)和巨细胞病毒(CMV))进行耐药性的遗传检测,但仍在探索WGS对其的应用。一项使用针对HIV的全基因组关联研究的研究能够准确地检测5种遗传关联,这些关联导至已知的氨基酸改变,从而赋予耐药性57。病毒WGS方法最大的潜在优势之一是能够检测出Sanger测序无法检测到的耐药亚群。一项研究表明,在HIV的检测中WGS可以提高检测HIV-1的低频耐药突变的敏感性58。WGS最令人兴奋的潜在应用之一是其预测抗药性(AMR)的能力,与传统的表型方法相比,它可以更快地提供初步结果。许多发表的报告显示了将WGS用作检测各种细菌耐药性的分子抗菌测试方法的希望,这表明耐药基因型与表型结果之间具有高度相关性。一项研究表明,针对15种抗菌药物测试的76株肠杆菌具有97.8%的特异性和99.6%的敏感性59。在其他测试肺炎克雷伯菌的研究中,观察到相似的敏感性和特异性(≥90%)60,61,铜绿假单胞菌62,金黄色葡萄球菌49和淋病奈瑟菌63。最近,已经基于WGS应用了机器学习模型来预测抗生素的最小抑菌浓度,非伤寒沙门氏菌的平均准确度为95%64,肺炎克雷伯菌为92%61。2.靶向测序在感染性疾病中的应用:tNGS在文库制备和测序之前使用选择过程来富集目标微生物序列。可以通过多种选择方法来实现富集,例如PCR扩增(通常称为扩增子测序),探针杂交以及利用CRISPR-Cas965,66。tNGS优于宏基因组学方法的优势在于,它克服了在大量细胞样本中扩增少量微生物序列的“大海捞针”的难题67。但是,富集过程(例如,针对特定基因的多重PCR)可能会导至结果偏倚。对于tNGS的临床应用,主要目标是鉴定患者样本中的一种或多种微生物病原体。但是,这些测定法也可能针对抗生素抗性基因。迄今为止,用于临床应用和微生物组研究的最普遍的富集方法是在NGS之前通过PCR扩增16S核糖体RNA(rRNA)基因65,68,69。但是,富集的替代方法也正在开发中70。Sabat及其同事开发了一种细菌tNGS分析方法,该方法可通过PCR扩增并对疑似尿路感染,血液培养阳性和骨科样本患者的尿液样本中的整个16S-23S rRNA区域进行测序。与常规培养,通过商业系统鉴定和16S Sanger测序相比,16S-23S tNGS分析可准确识别血液和尿液中的病原体,并显示出骨科样本中细菌病原体检出率的增加。除了单纯的tNGS分析外,还在开发更大的多重检测组合。已经描述了使用多重探针富集步骤的两种检测方法,用于血液和组织样品中的病毒(VirCapSeq-VERT)和细菌(BacCapSeq)检测71,72。两者都使用2到400万个探针池来分别选择性覆盖300多种细菌和200种病毒物种的微生物序列,包括前者的AMR标记和毒力决定因素。与特定的单重PCR相比,这些检测方法显示出相似的检出限。VirCapSeq-VERT已在临床上应用,将其与mNGS进行了比较,以确定来源不明的发热病原体73。尽管两种方法都可以鉴定出相同的物种,但与宏基因组方法相比,VirCapSeq具有更高的灵敏度。3.宏基因组测序在感染性疾病中的应用:mNGS是一种可以直接从患者标本中检测全部核酸的方法。与tNGS方法不同,该方法不会选择性地扩增特定靶标,因为样品中的所有核酸均会被并行扩增和测序,从而可以无偏的检测所有微生物组(即细菌,病毒,寄生虫和真菌),抗药性标记,毒力因子,甚至是与不同疾病状态相关的宿主生物标记。这提供了直接从患者样本中进行无假设诊断的优势。包括《中国宏基因组学第二代测序技术检测感染病原体的临床应用专家共识》74等在内的一系列共识已由国内专家提出,共识中指出了如何宿主基因干扰,如何判定是否是致病菌,如何解读结果等临床应用疑难点,表明宏基因组测序在临床感染性疾病中的应用需要在技术方法等层面进一步完善。方法学的变异包括基于DNA和RNA(也称为宏转录组学)的方法,可从感染源或无细胞DNA(cfDNA)中检测出完整的微生物。cfDNA(来自死亡的溶解的微生物的小核酸片段,被过滤到血液或尿液中)可以提供远端感染部位(例如,肺炎期间的肺部)的信息。重要的是要了解这些方法的差异和局限性。例如,如果在mNGS中未包含基于RNA的方法,则将无法检测RNA病毒,或者将无法基于转录组的分析来研究宿主的免疫反应75。mNGS方法学也是目前尚缺乏标准化的复杂,多步骤的过程,这也使结果解释变得更加复杂76。事实证明,mNGS作为诊断工具可成功检测出多个部位(包括中枢神经系统)的感染,血液,呼吸系统,胃肠道,假肢关节,尿路和眼部。确定的病原体包括细菌,分枝杆菌,RNA和DNA病毒,酵母和霉菌和寄生虫。在其中一些情况下,大量的标准诊断工具无法使用。mNGS在检测新型的,罕见的,和非典型原因或以前接受过治疗的患者方面特别成功。最近的一些研究表明,与护理标准诊断相比,mNGS的敏感性更高,对mNGS的前景具有指导意义。苗和同事的一项研究77观察了511种不同来源的标本的比较性能。回顾性图表显示,mNGS的整体临床敏感性和特异性分别为50.7%和85.7%,标准诊断法的整体临床敏感性和特异性分别为35.2%和89.1%。mNGS特别适用于结核分枝杆菌,病毒,厌氧菌和真菌。但是,同一项研究还表明,对于常见的细菌感染,尤其是在以前没有抗生素暴露的情况下,通过mNGS进行的检测并不优于培养。类似地,来自假体关节感染的超声液或滑液的mNGS在培养阴性病例中分别提供了25%和18.3%的增量产率78,79。另一种情况下,使用mNGS检查至今仍未明确诊断病原的94例亚急性或慢性脑膜炎患者的脑脊液,鉴定出2例猪带绦虫,1例HIV-1,4例真菌和1例蛛网膜下腔神经囊虫病80。这项研究表明,通常存在大量检测阴性病例81,但是,mNGS具有识别未知的病原体的潜力,并有助于疑难患者的治疗82,83。大多数最初的研究都集中在病原体检测上。但是,当覆盖范围足够大时,mNGS也可用于对一种或多种主要病原体进行菌株鉴定,或直接从标本中检测AMR和毒力因子基因。对于具有较小基因组的病原体,例如病毒则更为容易。例如,在随后的继发性非性传播病例中,对流行地区中与血液有关的寨卡病毒株进行分型84。最近应用CosmosID生物信息学平台查询mNGS结果葡萄球菌呈阳性,mecA介导的甲氧西林耐药性检测具有77.4%的敏感性和100%的特异性85。最后,可以查询基于RNA的方法中的宿主序列以研究免疫反应并将其整合到诊断算法中,以帮助确定微生物序列的重要性。令人印象深刻的是,Langelier和同事在下呼吸道感染中结合病原体,微生物组和宿主转录组分析,获得了100%的阴性预测值86。WGS在公共卫生实验室中应用广泛,有助于快速识别和跟踪传染病暴发,并检测新出现的耐药性和进行监测。tNGS在临床微生物学中未得到充分利用;然而,新的富集方法的发展将允许广泛的病原体检测和高灵敏度。将来,tNGS可能会成为一种更易于使用的检测方法。宏基因组下一代测序已成为一种有前途的单一、通用病原体检测(即细菌,真菌,寄生虫,病毒)方法,可直接从临床标本中进行传染病诊断。

