来源: 奥咨达医疗技术服务作者:2021年05月27日 09:39
新版欧盟《医疗器械法规(MDR)》落地后,对国内所有医疗器械出口企业来说,都将产生翻天覆地的变化!
得益于国家政策的扶持以及市场需求的增加,我国医疗器械行业销售规模近几年整体保持增长态势,医疗器械出口额度逐年增加:2019年出口总额达到891.29亿人民币,同比增长达13.13%;2020年医疗仪器及机械出口总额为1259.3亿人民币,同比增长41.5%。
但是,新版欧盟《医疗器械法规(MDR)》落地后,对国内所有医疗器械出口企业来说,都将产生翻天覆地的变化!

欧盟MDR法规,今日生效!
2017年5月5日,欧盟正式发布新版医疗器械法规MDR(EU 2017/745),取代旧的医疗器械指令MDD( 93/42/EEC)。新旧法规交替过渡期为三年。
2019年,海关总署组织技术性贸易措施研究评议基地成功应对欧盟MDR新规,促使欧盟在2020年4月24日正式宣布将《医疗器械法规(MDR)》强制实施日期推迟一年,这为中国医疗器械生产企业争取到了宝贵的缓冲时间,但强制执行日期为2021年5月26日,也就是昨天,《医疗器械法规(MDR)》正式落地!
新版法规,对械企的要求更高
根据欧盟新颁布的医疗器械法规《医疗器械法规》(2017/745,MDR)和《体外诊断器械法规》(2017/746,IVDR),欧盟将医疗器械分为两大类别:医疗器械MD和体外诊断器械IVD。
MDR法规执行时间为2021年5月26日,IVDR法规执行时间为2022年5月26日。所以,对于所有的体外诊断企业来说,还有一年的时间为欧盟的体外设备法规(IVDR)做准备。
据了解,MDR后侵入式医疗器械MD根据风险等级将再细分为I、IIa、 IIb、III类;非侵入式体外诊断器械IVD依据风险等级由低到高细分为A、B、C、D四类。
总体来说,欧盟MDR新规更加关注临床性能、更好的医疗器械可追溯性和对患者更大的透明度,具体变化有:医疗器械的范围扩大;提出医疗器械新概念和定义;设立中央电子资料库(Eudamed);设立产品独立的产品识别码(UDI);完善了医疗器械的通用安全和性能要求;加强对技术文件的要求;加强器械上市后的监管;完善临床评价相关要求;对授权认证机构(NB)提出严格要求等。
这意味着对进入欧洲市场的医疗器械将实施更严格的限制,对医疗器械相关企业提出了更高的要求。
80%中国械企或被迫放弃CE证书
毋庸置疑,MDR的落地将给所有中国出口械企带来了不少的麻烦,比如成本增加、认证周期拉长、合规风险增大。
粗看之下,CE流程变化不大,但其实细节上的变化非常大,包括各个方面如安全性能要求、临床期望、标签、规格书、上市后要求等,因此,所有器械都会受到一定的影响。
据悉,因为受到MDR的影响,德国图特林根地区行业协会预估当地大概会有100-200家小型企业面临倒闭或者转让。
更有业内人士预测,严格落地后的MDR将致使欧洲30%的医疗公司面临倒闭风险,超80%的中国企业被迫放弃CE证书......
在MDR实施之后,三年过渡期内仍然可以按照MDD和AIMDD申请CE证书并保持证书的有效性,根据规定,过渡期内NB签发的CE证书继续有效,但是从其交付日期起有效期不超过5年,并且于2024年5月27日失效。
所以,MDR落地后,对于已经获得CE证书的所有相关企业,第一件事就是要赶快重新确认产品的风险分类等级,确认好是否有风险等级升级的可能。并尽快确认原CE证书的发证机构是否已经获得了欧盟当局的批准,是否还具备MDR证书颁发的资格。
要知道,MDR之后,能力和经验将是审查的重要指标,相比MDD时期,MDR的第三方认证机构必然会少很多,所以企业必须尽快修改原CE技术文件,再次向具有MDR发证资质的机构提出新的认证申请,以获得MDR法规下的新CE证书。
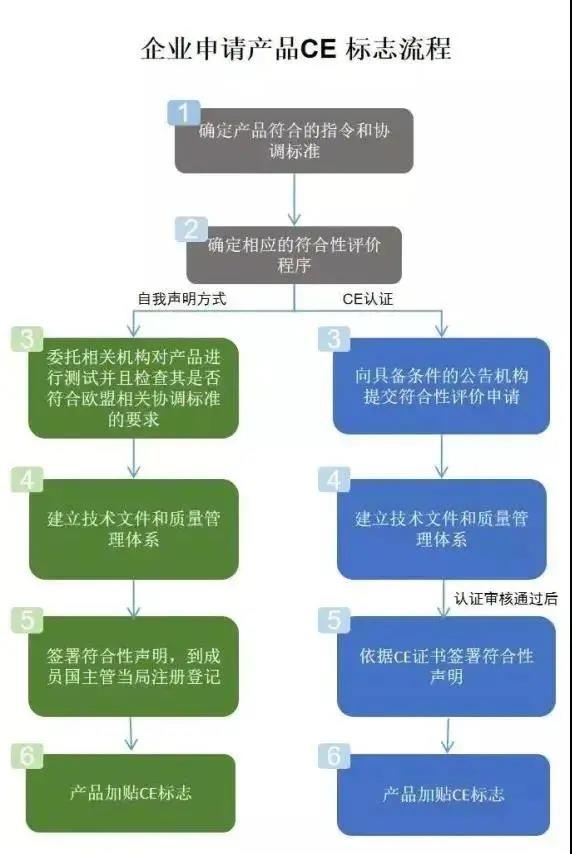
无疑,欧盟MDR新规的落地,使所有中国出口械企增加了更多的困难和风险,但面对海外市场相信没有企业会选择退缩。所以在此提醒所有企业一定要加强医疗器械质量意识和责任意识,按照MDR法规要求合规生产,保障产品和标准的符合性。
同时加强与国外客户之间的联系和沟通,明确生产标准和认证要求,避免后续认证和价格纠纷。
本文著作权属原创者所有,不代表本站立场。我们转载此文出于传播更多资讯之目的,如涉著作权事宜请联系删除。

