医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。根据商务部消息,RCEP即将于明年1月1日对文莱、柬埔寨、老挝、新加坡、泰国、越南、中国、日本、新西兰、澳大利亚10国生效。另据韩联社消息,RCEP目前已在韩国国会外交统一委员会待决。韩产业通商资源部近日表示:"我国也将于本月内在国会处理批准动议案后,于明年1月生效。
为帮助企业在RCEP协议生效后就可以合规高效地进行医疗器械进出口活动,我们在此汇总了RCEP成员国医疗器械准入程序及注册流程供企业参考。本篇我们主要汇总了:韩国、日本、澳大利亚、泰国、马来西亚对医疗器械产品的准入要求。下一期,我们将汇总其他成员国,敬请期待~
2、RCEP部分成员国医疗器械分类及要求
韩国KFDA
韩国卫生福利部,简称卫生部,主要负责管食品、药品、化妆品和医疗器械的管理,是最主要的卫生保健部门。
依照《医疗器械法》,韩国卫生福利部下属的食品药品安全部负责对医疗器械的监管工作。
韩国医疗器械法把医疗器械分为4类(Ⅰ、Ⅱ、Ⅲ、Ⅳ),从Ⅰ类至Ⅳ类风险程度递增,即:Ⅰ类为几乎没有潜在危险的医疗器械,Ⅳ类为高风险的医疗器械。
分类
Ⅰ类:几乎没有潜在危险的医疗器械;
Ⅱ类:具有低潜在危险的医疗器械;
Ⅲ类:具有中度潜在危险的医疗器械;
Ⅳ类:高风险的医疗器械。
注册流程
1、确定产品分类(I、II、III、IV),选择韩代KLH(韩国证书持证人,不在韩国境内的企业需要选择一个韩国证书持证人,通常为在韩国的分销商);
2、II类产品需申请KGMP证书和接受现场审核,II类产品一般是授权的第三方审核员,并获得KGMP证书;
3、II类产品需要送样品到韩国MFDS授权的实验室进行韩国标准的测试;
4、由韩代向MFDS提交技术文件(检测报告,KGMP证书等),进行注册审批;
5、支付申请费用;
6、注册文件整改,注册批准;
7、指定韩国代理商和经销商,产品销售。
日本
日本的医疗器械注册机构叫做PMDA,全程英文也就是Pharmaceuticals and Medical Devices Agency。
PMDA注册流程:
流程
第一步:准备阶段。确定产品分类(I,II特殊控制,II类控制,III,IV)和产品JMDN编码,选择日代MAH。1个月。
第二步:制造商向PMDA注册工厂,1个月。
第三步:II类特殊控制产品向授权认证机构PCB申请QMS工厂审核,其他II类产品和III类IV类产品向PMDA申请QMS工厂审核,并获得QMS证书,3个月。
第四步:申请Pre-Market Apporval证书,II类特殊控制由PCB发证,其他II类产品和III类IV类产品控制由MHLW发证。3个月。
第五步:支付申请费用。
第六步:注册文件整改,注册批准。
第七步:所有类别产品均需要MAH向RBHW进行进口通报注册后才能进口销售。
参考自网络
澳大利亚TGA
澳大利亚药品管理局(TGA),是澳洲医疗用品的监管机构,负责一系列评估和监管确保澳洲药品保质保量。TGA监管的产品范围包括药品,医疗器械,血液及血液产品。
分类
澳大利亚将医疗器械根据其构成的风险程度分为五大类。分类级别越高,要求就越苛刻。
准入程序
一、如果医疗器械是在海外制造的(即I类消毒; I类测量; IIa类; IIb类,III类,AIMD类)
1. 制造商从TGA或欧盟的认证机构获得合格评定证据;
2. 制造商准备澳大利亚符合性声明;
3. 主办者向TGA提交制造商的证据;
4. 主办者递交在ARTG登记申请;
5. 在ARTG登记后主办者可以在澳大利亚供应器械;
6. 器械上市后持续监控。
二、如果该器械包含一种药物或动物材料,微生物或人类起源物(即 III类和 AIMD类):
1. 制造商决定质量规程,用于证示器械符合相关的基本原则,并准备必要的文件;
2. 制造商申请TGA合格评定证;
3. 制造商准备澳大利亚符合性声明;
4. 主办者向TGA提交制造商的证据;
5. 主办者递交在ARTG登记申请;
6. 在ARTG登记后主办者可以在澳大利亚供应器械;
7. 器械上市后持续监视。
印度尼西亚
隶属于印度尼西亚卫生部(Ministry of Health Republic of Indonesia,MoH)的医药服务和医疗器械总局负责国产和进口医疗器械的法规性事务,卫生部则对医疗器械的销售进行管理。印尼卫生部规定,在印尼销售医疗器械(包含进口产品在内)的公司必需是当地实体公司,且必需拥有印尼卫生部颁发的医疗器械经营授权书;同时规定外国企业必需有当地经销商或者在当地设办事处;印尼卫生部禁止进口二手设备。
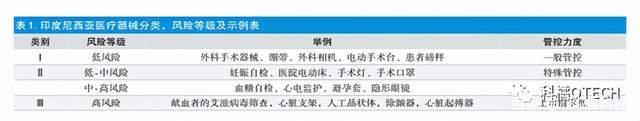
印尼卫部按照风险等级高低及管控力度的强弱将医疗器械分为3类:Ⅰ类:低风险器械—一般管控;Ⅱ类:中级风险器械—特殊管控;Ⅲ类:高风险器械—上市前审批。
分类
Ⅰ类:低风险器械—一般管控;
Ⅱ类:中级风险器械—特殊管控;
Ⅲ类:高风险器械—上市前审批。
注册流程
印尼卫生部规定所有进口医疗器械都需要进行注册审批。值得关注的是,印尼政府为更好融入东南亚国家联盟法规体系,支持“在线”提交注册申报资料。进口医疗器械委托持有印尼卫生部颁发的医疗器械经营授权书的实体公司在印尼国家网站(
http://www.regalkes.depkes.go.id)申请生产、经销执照或注册产品。
印尼大使馆是医疗器械入市的头道“关卡”—实体代理商需要获得由印尼大使馆颁发的经营授权书(Letter of Authority,LOA),同时制造商需要出具产品确认信。
获得经营授权书后,进口商需要向医疗器械主管部门—医药服务和医疗器械总局提交如下文件:
①自由销售证明;
②既往销售国清单;
③已获得的医疗器械注册证、CE或FDA认证(若有会明显加快审评审批的进度);
④产品本身的介绍(包含预期用途、适应症、禁忌症、预防措施及潜在的危害等);
⑤生产工艺及质量控制报告(有源医疗器械需要提供符合IEC 60601或IEC 61010要求的检测报告);
⑥使用说明书;
⑦产品包装形式(需要以照片形式提供前视图和后视图)以及对产品编码或系列号的解释;
⑧产品包含内容的清单,包括组件等(需以照片形式提供);
⑨印尼对进口产品的本地测试不做要求,认可国外的临床评价资料[7]。
但有5类产品在注册时被特殊要求:
①艾滋病毒相关产品(HIV Products):需在印尼国家实验室医院(Indonesia Reference National Laboratory Hospital,RSCM)检测;
②月经垫、成人尿布、避孕套、注射器:需在印度尼西亚国家实验室进行荧光检测;
③产品原材料含动物来源的成分(如羊肠线):需提供原产国的免疫证明;
④产品辐射(例如:X射线机):需提供国家原子能机构的辐射安全证书;
⑤开放软件:需提供制造商或独立实验室的软件验证报告。
印尼政府为进口医疗企业颁发的注册证效期为5年,与我国类似,在注册证到期前至少6个月提交延续注册申请;若生产地址、商品名、适用范围、证书持有人发生变化需要新注册;若新增型号、标签说明书的微小变更及灭菌场地和/或技术指标变化需进行变更注册。
参考自:《“医疗旅游”背景下泰国和印度尼西亚医疗器械市场准入要求》 作者张坤智等。
泰国
泰国公共卫生部下属的泰国食品和药物管理局(Thailand Food and Drug Administration TFDA)负责监管本国医疗器械。当前的泰国医疗器械监管制度自2008年起开始实施,制度规定,所有的国产及进口医疗器械均需经注册后方可进入本国市场。医疗器械控制部门(Medical Device Control Division)作为TFDA的下属机构,专门负责监督医疗器械的法规相关事务。TFDA禁止进口二手/翻新的设备,进口产品在原产国必需取得自由销售证明(Certificate of Free Sale)。
TFDA对医疗器械的分类区别于绝大多数国家,分类如下:
分类
需获得许可的医疗器械,等级最高(Licensed Medical Device,Class I);
需获得通知的医疗器械,等级次之(Notification Medical Device,Class Ⅱ);
通用医疗器械,等级最低(General Medical Device,Class Ⅲ)
备注:
在Class I(需获得许可的医疗器械)这个管控最严格的类别中,目前有7种设备,均需经过TFDA的审批:①避孕套;②医用检查手套;③外科手套;④无菌一次性注射器;⑤无菌一次性胰岛素注射器;⑥用于诊断的艾滋病毒检测试剂;⑦隐形眼镜。
Class Ⅱ(需获得通知的医疗器械)需要生产商和进口商在进口入泰国前,从TFDA处获得产品的通知,含以下设备:①物理理疗设备;②酒精检测仪;③乳房植入硅胶假体;④丰胸产品;⑤艾滋病毒检测试剂盒(仅限研究用)。
未在Class Ⅰ、Class Ⅱ中的其他医疗器械为Class Ⅲ(通用医疗器械),Class Ⅲ覆盖了90%左右的医疗器械
特别提醒:
值得关注的是,若医疗器械涵盖化妆品或者药物成分,申请人可以要求TFDA对其属性及风险等级进行评价,进而确定产品是否划分为医疗器械。(泰国卫生部2019年将医疗器械法案升级,过氧化氢浓度高于6%的牙齿美白产品、过氧化脲或其他过氧化氢存在或释放至6%的产品被界定为“需获得通知的医疗器械”;升级的法案将隐形眼镜护理产品也定义为医疗器械且被分类为“需获得通知的医疗器械”)
注册流程
医疗器械在泰国的注册流程与我国医疗器械注册过程有颇多相似之处:
①申请者准备注册所需的相关文件,并且保证自由销售证明(Certificate of Free Sale)和体系证明合规;
②申请者将注册所需的文件提交至TFDA服务中心;
③提交的文件由医疗设备控制部门的监管人员进行审核和记录;
④工作人员将申请人的档案记录到数据库系统中,随后向申请人提供一个参考编号和收据;
⑤若监管人员核实所提交的文件均属正确,并符合相关标准,便会在自由销售证明的背面印上医疗仪器进口通知书。监管机构随后将这些文件提交给医疗设备控制部门的负责人进行最后批准;
⑥申请者最终取得进口批准书。完成注册流程的耗时取决于申请材料合规程度。与我国医疗器械注册审评的补正环节类似,若申请材料不完整或不符合TFDA规定,监管机构有权要求申请人在修订或补充后重新提交申请资料。
与新加坡、马来西亚等国对于临床试验的规定不同,在泰国注册的进口医疗器械通常不需要临床实验研究,也不需要在泰国本地进行类型检测或样本测试,但所有进口医疗器械进入泰国市场之前都需要获得原产国的注册批准。
根据进口疗器械的分类不同,注册所需的文件清单也有所差异。
Class I—需获得许可的医疗器械注册所需文件:①自由销售证明;②质量安全证明,如ISO证书;③临床评估报告;④无菌测试证明(如适用);⑤稳定性证明;⑥原材料及成品说明书;⑦泰语宣传页、产品照片;⑧生产过程说明;⑨TFDA要求申请人使用东盟文件提交档案模板(ASEAN Common Submission Dossier Template,CSDT)提交医疗器械相关的文档。
Class Ⅱ—需获得通知的医疗器械注册所需文件:①申请信;②申请表;③文件清单;④所申请医疗器械的结构、材料等特征;⑤包装信息;⑥组成数量;⑦预期用途、警告及注意事项;⑧使用说明;⑨贮存条件;⑩性能检测报告;自由销售证明;营业执照复印件及申请人的身份证复印件;使用说明书。
Class Ⅲ—通用医疗器械注册所需文件:①公司请求TFDA验证其自由销售证明的函件;②制造商的两份自由销售证明和质量体系证书;③代理公司注册文件及经营目标;④所有的进口通用医疗器械都需要随附授权书和当地代理商ID卡的认证副本;⑤自由销售证明中指定的产品目录或产品详细信息,需显示医疗设备的代码(TFDA手册中指出了与医疗设备类型相关的各种代码);⑥代理公司的进口许可证副本;⑦可能需要有关医疗设备安全性或预期用途的其他文档。
泰国政府为进口Class Ⅱ和Class Ⅲ类医疗企业颁发的注册证效期为5年,Class Ⅰ类医疗器械的有效期则视自由销售证明的有效期而定。
参考自:《“医疗旅游”背景下泰国和印度尼西亚医疗器械市场准入要求》作者张坤智等
马来西亚
马来西亚医疗器械监管的主管部门为卫生部下属的医疗器械管理局(Medi-cal Device Athority,MDA),主要职责范围是:责成医疗器械厂商(本国或外国)严格遵守《医疗器械质量管理体系》(ISO13485)的质量体系要求;指导厂商进行创新产品的临床试验;对本国及进口的医疗器械产品进行注册及疏通医疗器械国内销售渠道。
2013 版马来西亚《医疗器械管理法》根据风险高低将医疗器械划分为 A、B、C、D 4 类:A 类医疗器械风险最低,B 类、C 类居中,D 类风险最高[7-8]。各个类别风险程度及产品举例详见下表:
分类
A 类:医疗器械风险最低;
B、C类:风险居中;
D类:风险最高。

注册流程
按照马来西亚《医疗器械管理法》规定,所有在马来西亚生产、进口的医疗器械产品都必须在医疗器械管理局进行注册后方可进入市场。对于进口的医疗器械生产商而言,需找到授权代理(authorizedrepresentative,AR)完成注册事宜。进口医疗器械经过如单独/组合使用医疗器械、成系列使用医疗器械等大致分组之后,根据风险类别高低,相应的注册流程有所差异 :
体外诊断试剂的分类及注册:
体外诊断试剂(in-vitro diagnostic,IVD)是指仅仅或主要用来为人体提供诊断、监测信息而单独或组合使用的装置,包括试剂、校准器、样品容器等[11]。《医疗器械管理法》根据对个人及公众健康风险影响程度的高低将 IVD 产品划分为 A、B、C、D 4 类[11],各个类别风险程度及产品举例详见下表:

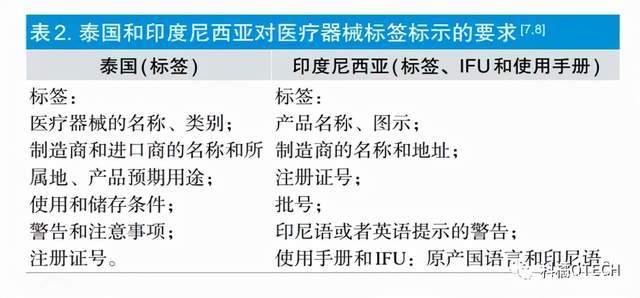
医疗器械标签标识的规定
首先是“三严禁”和“六必须”。“三严禁”:严禁出售任何未贴标签的医疗器械、严禁操作任何未附标签的医疗器械以及严禁调查测试任何未附标签的医疗器械。“六必须”:包含进口产品在内的注册医疗器械标签上必须注明马来西亚医疗器械注册号码;标签中必须删去任何直接或间接表示该产品功效由卫生部或其他组织推广及认可的描述;医疗器械的标签必须清晰、持久且放置于显著的位置;标签的格式、内容及可读性必须与其配套设备、预期用途和预期用户的技术、经验、教育水平相匹配,适当的情况下可辅以图纸和图表;所有的纸质标签都必须附在产品上;风险分析过程中发现的任何残留风险必须在标签中明确反映为禁忌证或警告。标签可通过例如印刷文件、电子显示屏等多种媒介向用户提供,且标签可以在进后或生产后制作,并需在医疗器械注册时提交。其次,家用医疗器械的标签必须使用马来西亚语,非家用医疗器械标签必须使用英语,必要时也可使用其他语种。在标签中鼓励使用国际认可的标志,同时在提供明确解释的情况下不排斥引入新符号。最后,标签标识中至少需要出现以下内容:规格型号及批号;制造商名称和联系方式;与设备相关的技术细节和预期用途;使用说明;使用过程中可能出现的不良反应、警告及/或预防措施;上市后的服务;使用后的报废方式。此外,医疗器械的贮存条件、使用前消毒灭菌程序、限制重复使用次数等信息以及“一次性使用”“仅限研究用”“翻新器械”等标识可酌情添加。
可豁免注册的医疗器械
虽然《医疗器械管理法》规定所有的医疗器械都需经过注册才能上市,但有些医疗器械使用风险极低,或者是有资质的医生为患者专门定制,或仅供研究用。因此,医疗器械管理局在 2014 年第二次会议上讨论对“低风险、限制使用且可由通知的方式加以控制”的医疗器械实行豁免注册。这类器械包括:低风险的医疗器械;有资质的医护人员为患者专门定制,且运用于突发情况或传统的救治方法无效的环境中的医疗器械;仅供临床研究及教学展示用的医疗器械。尽管上述医疗器械可被豁免注册,但医疗器械管理局对其并未放松监管。上述医疗器械在进出口或上市前仍然必须接受医疗器械管理局的通告和督查。
参考自《浅析马来西亚医疗器械市场准入要求》作者张坤智等


